
合肥医药厂制剂车间洁净厂房设计验收方案
来源:www.kqhjh.com 作者:空气好净化
发布时间:2021-08-12 09:04:07点击:次
合肥医药厂制剂车间洁净厂房主要是药品生产过程有直接联系的HVAC系统、水系统、工艺气体过滤系统、消防系统、臭氧消毒系统。新建制剂厂房各系统安装均按相关规范要求进行。

一、制剂车间洁净厂房总体布局设计
1、口服固体制剂生产车间厂房在锅炉房的上风口处,主要系统包括空调机房、制水站、空压机房、配电室等。
2、口服固体制剂生产车间车间内部墙体及吊顶均为彩钢结构,用中性密封胶密封缝隙;不锈钢圆弧进行衔接和密封,一般生产区全部为水磨石地面,净化区域为绿色环保环氧树脂自流平地坪。门框和密封窗框均采用异形铝材,洁净区安全门为正面玻璃—临时破开结构,在明显位置设有破门工具。主要管道设置于技术夹层,并对连接部位进行密封。
3、口服固体制剂生产车间洁净厂房结构围护、隔断均为δ=50mm岩棉夹芯彩钢板,顶面与墙面、围护墙上下角、地面与围护墙面之间的夹角均为R50铝合金弧形连接,对角为三维角结构,玻璃与夹芯彩钢板之间过度为圆坡形,易于清洁。
4、口服固体制剂洁净区应包括:物料传递室、原辅料暂存室、粉碎室、称量备料室及前室、制粒干燥室、研磨热融室、挤出滚圆室、总混室、压片室、胶囊充填室、流、中转站、铝塑包装室、塑瓶包装间、洁具室、器具清洗室、器具存放室、IPC室、废弃物传递室以及人员洗手消毒、手消毒,洁净级别设置为D级。
5、根据新版GMP(2010年修订)要求,洁净区净化送风采用顶送下侧回风形式;粉碎室、制粒干燥室、总混室、称量备料室等相对负压房间采用侧墙直排。洁净区送风终端均采用高效过滤,回风口为可拆洗铝合金百页窗(过滤层为纱网无纺布),回风口底边距地面200mm,送风系统管道采用优质镀锌板制作,回风采用角柱形竖井回风,对直排管道加装止回阀。
6、不同等级的洁净室之间的压差大于10Pa,洁净室与外环境压差大于10Pa,并装有微压差计。为保证车间空调系统的正常使用,空调送风系统设置有自动故障报警系统。
7、配电系统、工艺用水系统及物料系统走技术夹层,车间垂直管路供给。洁净区物料供给管道全部采用304不锈钢材质,夹层与洁净区之间管道按要求进行密封,并注塑进行密封保护,以实施有效的隔离。洁净区地漏全部用水封式地漏。
8、为了保证生产车间的消防安全,车间设有安全防火烟感自动报警系统,并配置有消防栓和灭火器,及自动抽烟系统。一般生产区配备的干粉灭火器,洁净区配备的气体灭火器。
9、由于产品生产对洁净室的环境温度和相对湿度没有特殊的要求,因此对洁净区的温湿度进行整体的控制,系统送风和回风均设置有温湿度显示仪,有效的控制送风温湿度,和监控回风温湿度,便于对车间洁净区温湿度进行整体的控制和调整。
10、车间设有压缩空气系统用于药品生产和启动气动阀门开关等,直接接触药品生产的压缩空气经除油、除水、除菌三级过滤,三级过滤后的压缩空气管道全部采用316L的不锈钢材质制作。
11、车间人流、物流分开设置,进入洁净区的人流通道、物流通道不产生交叉污染,人员净化更及后进入洁净区,物料外清后,经物料传送室进入洁净区。

二、制剂净化车间厂房确认目的
确认制剂净化车间厂房及设施设计及建设符合《医药工业洁净厂房设计规范》(GB50457-2008)规范要求,空气净化系统设计、安装符合《药品生产质量管理规范》(2010年修订)要求,能够正常运行,各项性能指标符合设计要求,确保制剂车间空气净化系统符合规范要求。主要依据如下:
1、《药品生产质量管理规范》(2010年修订);
2、空调净化系统操作及设备说明书等技术资料;
3、《药品生产确认指南》(2003);
4、《新版GMP实施指南》;
5、《确认管理规程》;
6、《医药工业洁净室悬浮粒子检测方法》(GB/T16292-2010);
7、《医药工业洁净室(区)浮游菌测试方法》(GB/T16293-2010);
8、《医药工业洁净室(区)沉降菌的测试方法》(GB/T16294-2010);
9、《洁净室及相关受控环境国际标准》(ISO14644.1~7);
10、《建筑给水排水设计规范》(GB50015-2003);
11、《建筑灭火器配制设计规范》(GB50140-2005);
12、《建筑设计防火规范》(GB50016-2006);
13、《建筑地面设计规范》(GB50037-2002);
14、《砌体结构设计规范》(GB50003-2001);
15、《医药工业洁净厂房设计规范》(GB50457-2008);
16、《洁净室施工及验收规范》(GB50591-2010)。

三、确认内容
1、依据《洁净室施工及验收规范》(GB50591-2010)确认厂房及设施符合医药工业洁净厂房设计规范》(GB50457-2008)要求,确定制剂厂房及设施与空调净化系统的技术指标及其设计要求符合设计要求,确认系统材质和质量符合预定要求及GMP规范的要求,系统的设计标准能满足制剂车间洁净度的要求。
2、制剂车间洁净厂房及设施是保证空调净化系统完整实现的基础与平台,消防设施是安全生产的保证,空调净化系统是保障生产洁净环境的关键设施。净化工程有限公司按《药品生产质量管理规范》(2010修订)的标准和双方确定的工艺布局进行设计、制造、安装空调净化系统。空调净化系统设有冷水机组、凉水塔、组合式空调机组、送回风管道、高效风口、回风口、新风口。管道系统有消声器、防火阀、风量调节阀。
3、性能确认内容:洁净室(区)换气次数、温湿度、压差、悬浮粒子、沉降菌、浮游菌、高效过滤器的风速、风量、噪声、照度。高效送风口风速及换气次数测试。
4、高效送风口测试目的:是通过风速的测试计算各房间实际风量,再根据各房间实际风量和体积计算出换气次数,证明系统的换气次数符合洁净区设计要求。若有偏差,应调节相应的风阀使系统符合设计标准要求。
5、智能风速计测试方法:按下图指示位置在15分钟内对准高效送风口过滤器下迎气流方向测试5点风速,计算平均风速。可据风口形状、大小来设计测风点数。
6、标准:洁净级别在C级或D万级的风口测试风量与设计风量之差应在设计风量的±15%内(换气次数:D级(≥18次/h)、C级(≥25次/h)、A级单向层流应均匀送风,风速为0.36~0.54m/s(指导值)(A级只测风速)。
7、房间静压差测试:测试仪器为压差计,压差应在风量测试之后进行,并应从平面上最里面的房间依次向外测试。测量前应将所有的门都关闭,测量时不允许有人穿越房间。标准为生产运行时,保证空气洁净级别不同的相邻房间之间静压差应大于10帕;洁净室(区)与室外大气静压差应大于10帕;同一空气洁净级别的相邻房间之间静压差应大于10帕生产结束后,保证空气洁净级别不同的相邻房间之间静压差应大于5帕;洁净室(区)与室外大气静压差应大于5帕;同一空气洁净级别的相邻房间之间静压差应大于2帕。
8、房间温湿度测试:测试仪器为温湿度计,主要于空调净化系统运行1小时后,用温湿度压差测试仪分别测试各房间温度和湿度,共测3天进行,每天测一次,并记录每次测试结果。要求温、湿度测试应在风量风压调整后进行。测点应放在洁净室有代表性的工作区或洁净室中心。标准为温度:18~26℃;相对湿度:45~65%。
9、为使药品生产环境达到规定空气净化级别要求,需对洁净区空气做灭菌处理,我们现采用的方法为臭氧灭菌处理。根据GMP要求必须对灭菌效果进行检测并确认。臭氧在常温、常压下分子结构不稳定,很快自行分解成氧(O2)和单个氧原子(O),后者具有很强的活性,对细菌有极强的氧化作用,臭氧氧化分解了细菌内部氧化葡萄糖所必需的酸,从而破坏其细胞膜,将它杀死。多余的氧原子则会自行重新组合成为普氧分子(O2),不存在任何有毒残留物,故称无污染消毒剂,它不但对各种细菌有较强的杀灭能力,而且对杀死霉菌也很有效。
臭氧浓度的测试方法:拟定在臭氧发生器开启20分钟后,每隔10分钟检测人员进入具有代表性的洁净房间,用臭氧气体检测仪测试室内臭氧浓度;根据卫生部《消毒技术规范》记载,臭氧对空气中的微生物有明显的杀灭作用,采用20mg/m3浓度的臭氧,作用30分钟,对自然菌的杀灭率达到90%以上。
通过公式计算,该洁净区臭氧达到消毒浓度时需要约12.6分钟,即可达到杀灭沉降菌所需臭氧浓度,为确保消毒效果,将臭氧消毒时间暂定为20分钟。可接受的标准:时间:开机20分钟后。保持运行30分钟。标准:净化区臭氧浓度≥10ppm。
10、洁净度级别相符认定条件:在静态情况下,对制剂生产车间进行功能间洁净度的相符性级别认定。认定标准为ISO14644-1。 相符性认定办法及依据ISO14644-1,对洁净区悬浮粒子进行测试,依据测试结果,对洁净度级别相符性认定,确认洁净区级别符合设计要求。D级洁净区(静态)空气悬浮粒子的级别分别为ISO 8。在确认级别时,应当使用采样管较短的便携式尘埃粒子计数器,避免≥5.0um悬浮粒子在远程采样系统的长采样管中沉降。在单向流系统中,应当采用等动力学的取样头。测试仪器为尘埃粒子计数器;测试部位为空调净化系统送风口、各级洁净室。
相符性认定办法及依据ISO14644-1,对洁净区悬浮粒子进行测试,依据测试结果,对洁净度级别相符性认定,确认洁净区级别符合设计要求。D级洁净区(静态)空气悬浮粒子的级别分别为ISO 8。在确认级别时,应当使用采样管较短的便携式尘埃粒子计数器,避免≥5.0um悬浮粒子在远程采样系统的长采样管中沉降。在单向流系统中,应当采用等动力学的取样头。测试仪器为尘埃粒子计数器;测试部位为空调净化系统送风口、各级洁净室。
相符性认定办法及依据ISO14644-1,对洁净区悬浮粒子进行测试,依据测试结果,对洁净度级别相符性认定,确认洁净区级别符合设计要求。D级洁净区(静态)空气悬浮粒子的级别分别为ISO 8。在确认级别时,应当使用采样管较短的便携式尘埃粒子计数器,避免≥5.0um悬浮粒子在远程采样系统的长采样管中沉降。在单向流系统中,应当采用等动力学的取样头。测试仪器为尘埃粒子计数器;测试部位为空调净化系统送风口、各级洁净室。
测试步骤:要在洁净室(区)净化空气系统已处于正常运行状态,洁净室(区)应没有生产人员的情况下进行测试。按照尘埃粒子计数器标准操作规程测量大于等于0.5um、大于等于5um粒子浓度。测量是静态测试,要在HVAC系统至少运行30分钟后开始采样,按照少量采样点数在房间内均匀分布,总采样次数不得少于3次。
11、采样点一般在离地面0.8m高度水平面上均匀布置,特殊情况下,可以就近选择,但原则上要避开回风口。采样点多于5点时,也可以在离地平0.8m~1.5m高度的区域内分层布置,但每层不少于5点。 采样次数的限定对于任何小洁净室或局部空气净化区域,采样点的数目不得少于2个,总采样次数不得少于5次。每个采样点的采样次数可以多于1次,且不同采样点的采样次数可以不同。
采样次数的限定对于任何小洁净室或局部空气净化区域,采样点的数目不得少于2个,总采样次数不得少于5次。每个采样点的采样次数可以多于1次,且不同采样点的采样次数可以不同。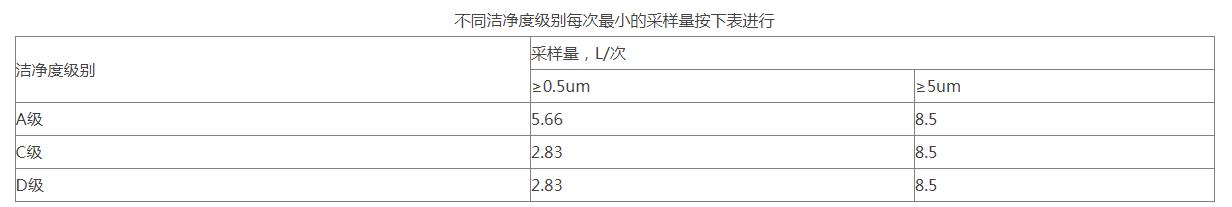 12、洁净度静态测试:在生产操作全部结束以后,全部操作人员撤出生产现场,经20分钟的自净后,进行空气悬浮粒子的静态检测,并确定警戒限和纠偏限(分别暂定为法定标准的50%及80%)。制剂车间的洁净级别为D级,在静态情况下,静态的标准为:
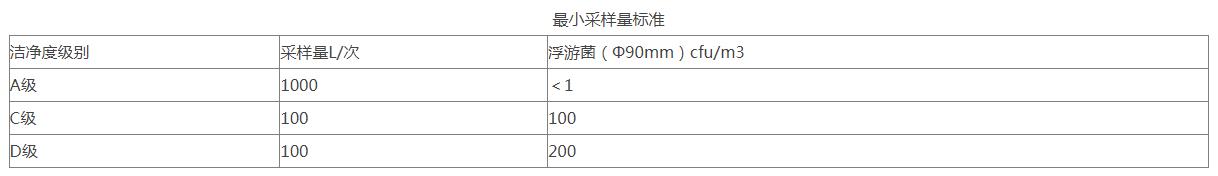
12、洁净度静态测试:在生产操作全部结束以后,全部操作人员撤出生产现场,经20分钟的自净后,进行空气悬浮粒子的静态检测,并确定警戒限和纠偏限(分别暂定为法定标准的50%及80%)。制剂车间的洁净级别为D级,在静态情况下,静态的标准为: 各洁净室沉降菌测试目的是确认空调净化系统的环境能够达到要求,并确定沉降菌的警戒限度和纠偏限度(分别暂定为法定标准的50%及80%)。主要测试方法为《医药工业洁净室(区)沉降菌的测试方法》(GB/T16294-2010);取样部位各洁净室房间。取样器具为培养皿(Φ90mm×15mm);取样步骤及方法在HVAC系统至少运行30分钟,房间的温湿度及相对压差达到要求后,方可进行沉降菌测试。用培养皿(Φ90mm×15mm)和琼脂培养基进行培养,在采样点放置,打开平皿盖,使培养基暴露时间可以少于4小时,将平皿盖盖上,然后在30~35℃条件下培养48小时后计数。(采样点数目同悬浮粒子)
各洁净室沉降菌测试目的是确认空调净化系统的环境能够达到要求,并确定沉降菌的警戒限度和纠偏限度(分别暂定为法定标准的50%及80%)。主要测试方法为《医药工业洁净室(区)沉降菌的测试方法》(GB/T16294-2010);取样部位各洁净室房间。取样器具为培养皿(Φ90mm×15mm);取样步骤及方法在HVAC系统至少运行30分钟,房间的温湿度及相对压差达到要求后,方可进行沉降菌测试。用培养皿(Φ90mm×15mm)和琼脂培养基进行培养,在采样点放置,打开平皿盖,使培养基暴露时间可以少于4小时,将平皿盖盖上,然后在30~35℃条件下培养48小时后计数。(采样点数目同悬浮粒子) 13、各洁净室浮游菌测试目的是确认空调净化系统的环境能够达到要求,并确定浮游菌的警戒限和纠偏限(分别暂定为法定标准的50%及80%)。测试依据《医药工业洁净室(区)浮游菌测试方法》(GB/T16293-2010);取样部位各洁净室房间。取样器具为Φ90mm玻璃平皿。取样步骤在HVAC系统至少运行30分钟,房间的温湿度及相对压差达到要求后,方可进行浮游菌菌测试。用Φ90mm玻璃平皿和大豆酪蛋白琼脂培养基(TSA)进行培养,在采样点放置,打开平皿盖,使培养基表面显露30分钟后,将平皿盖盖上,然后在30~35℃条件下培养48小时后计数。采样点的位置及采样点的数量可以同悬浮粒子测试要求。(采样点数目同悬浮粒子)每个采样点取样一次。(每个采样点一般只采样一次)
13、各洁净室浮游菌测试目的是确认空调净化系统的环境能够达到要求,并确定浮游菌的警戒限和纠偏限(分别暂定为法定标准的50%及80%)。测试依据《医药工业洁净室(区)浮游菌测试方法》(GB/T16293-2010);取样部位各洁净室房间。取样器具为Φ90mm玻璃平皿。取样步骤在HVAC系统至少运行30分钟,房间的温湿度及相对压差达到要求后,方可进行浮游菌菌测试。用Φ90mm玻璃平皿和大豆酪蛋白琼脂培养基(TSA)进行培养,在采样点放置,打开平皿盖,使培养基表面显露30分钟后,将平皿盖盖上,然后在30~35℃条件下培养48小时后计数。采样点的位置及采样点的数量可以同悬浮粒子测试要求。(采样点数目同悬浮粒子)每个采样点取样一次。(每个采样点一般只采样一次)
 14、各洁净室噪声、照度测试采用=噪声监测仪和照度计;通过用噪声监测仪及照度计在各主要功能房间进行测试。标准为噪声:≤75dB;照度:主要工作室照度应不低于300勒克斯,其他房间应不低于150勒克斯。
14、各洁净室噪声、照度测试采用=噪声监测仪和照度计;通过用噪声监测仪及照度计在各主要功能房间进行测试。标准为噪声:≤75dB;照度:主要工作室照度应不低于300勒克斯,其他房间应不低于150勒克斯。
采样次数的限定对于任何小洁净室或局部空气净化区域,采样点的数目不得少于2个,总采样次数不得少于5次。每个采样点的采样次数可以多于1次,且不同采样点的采样次数可以不同。 12、洁净度静态测试:在生产操作全部结束以后,全部操作人员撤出生产现场,经20分钟的自净后,进行空气悬浮粒子的静态检测,并确定警戒限和纠偏限(分别暂定为法定标准的50%及80%)。制剂车间的洁净级别为D级,在静态情况下,静态的标准为: 各洁净室沉降菌测试目的是确认空调净化系统的环境能够达到要求,并确定沉降菌的警戒限度和纠偏限度(分别暂定为法定标准的50%及80%)。主要测试方法为《医药工业洁净室(区)沉降菌的测试方法》(GB/T16294-2010);取样部位各洁净室房间。取样器具为培养皿(Φ90mm×15mm);取样步骤及方法在HVAC系统至少运行30分钟,房间的温湿度及相对压差达到要求后,方可进行沉降菌测试。用培养皿(Φ90mm×15mm)和琼脂培养基进行培养,在采样点放置,打开平皿盖,使培养基暴露时间可以少于4小时,将平皿盖盖上,然后在30~35℃条件下培养48小时后计数。(采样点数目同悬浮粒子) 13、各洁净室浮游菌测试目的是确认空调净化系统的环境能够达到要求,并确定浮游菌的警戒限和纠偏限(分别暂定为法定标准的50%及80%)。测试依据《医药工业洁净室(区)浮游菌测试方法》(GB/T16293-2010);取样部位各洁净室房间。取样器具为Φ90mm玻璃平皿。取样步骤在HVAC系统至少运行30分钟,房间的温湿度及相对压差达到要求后,方可进行浮游菌菌测试。用Φ90mm玻璃平皿和大豆酪蛋白琼脂培养基(TSA)进行培养,在采样点放置,打开平皿盖,使培养基表面显露30分钟后,将平皿盖盖上,然后在30~35℃条件下培养48小时后计数。采样点的位置及采样点的数量可以同悬浮粒子测试要求。(采样点数目同悬浮粒子)每个采样点取样一次。(每个采样点一般只采样一次) 14、各洁净室噪声、照度测试采用=噪声监测仪和照度计;通过用噪声监测仪及照度计在各主要功能房间进行测试。标准为噪声:≤75dB;照度:主要工作室照度应不低于300勒克斯,其他房间应不低于150勒克斯。
15、异常情况处理:系统运行确认过程中,应严格按照各标准操作程序、维护保养程序、设计要求进行操作和判定。出现个别项目不符合标准的结果时,应按下列程序进行处理:洁净室空气环境监测出现个别房间个别指标不合格的则应重新取样一次,检测不合格的指标;其它运行确认项目不符合要求时,应重新调试或请供应商现场调试;若属设备方面的原因,必要时报确认小组,调整设备运行参数或对设备进行处理。 监测依据:《洁净监测管理规程》(JH-GL-ZL-034-ROO);《洁净区沉降菌检测操作规程》(JH-CZ-ZL-130-ROO);《洁净区浮游菌检测操作规程》(JH-CZ-ZL-146-ROO);《洁净区尘埃粒子检测操作规程》(JH-CZ-ZL-131-ROO);《洁净区风速、风量、换气次数测试操作规程》(JH-CZ-ZL-209-ROO)。
监测依据:《洁净监测管理规程》(JH-GL-ZL-034-ROO);《洁净区沉降菌检测操作规程》(JH-CZ-ZL-130-ROO);《洁净区浮游菌检测操作规程》(JH-CZ-ZL-146-ROO);《洁净区尘埃粒子检测操作规程》(JH-CZ-ZL-131-ROO);《洁净区风速、风量、换气次数测试操作规程》(JH-CZ-ZL-209-ROO)。
监测依据:《洁净监测管理规程》(JH-GL-ZL-034-ROO);《洁净区沉降菌检测操作规程》(JH-CZ-ZL-130-ROO);《洁净区浮游菌检测操作规程》(JH-CZ-ZL-146-ROO);《洁净区尘埃粒子检测操作规程》(JH-CZ-ZL-131-ROO);《洁净区风速、风量、换气次数测试操作规程》(JH-CZ-ZL-209-ROO)。
16、偏差处理:在执行本方案过程中发生任何偏差均应有文件记录,并将所有偏差情况描述清楚,并列出偏差纠正结果,说明引起偏差的原因。实施责任人应保证结论正确无误。确认小组应对本系统中所有偏差与纠正予以审核、认可。
安徽空气好净化工程有限公司是一家专注无尘车间、GMP车间、无菌车间、洁净实验室、医疗手术室等净化工程洁净室系统研发、设计、施工、升级、售后为一体的净化工程全案解决方案服务商。立足安徽,服务全国,高标准要求工程质量,为客户提供高品质的客户服务,整体改善净化洁净室环境,打造国内安全舒适的实验室手术室环境,精品品牌造就精品工程,矢志成为中国净化工程行业精品品牌。